
The Role Quality Plays in Medical Device Creation
Medical Device startups working on their first product launch have a steep hill to climb. In addition to typical considerations like funding, marketing, sales, and product features, medical device startups also have to take regulatory and quality pathways into account, as well as building a robust quality management system (QMS). Medical device startups often focus so much of their time on the regulatory pathway that they neglect to plan and fund their QMS, but you'll discover that it's never too early to start planning for it.
Because standards vary from country to country, learning how to navigate specific regulatory and quality pathways is crucial, as each device will have different needs and endpoints. For this article, we'll focus mainly on US requirements for medical device manufacturers, but the good news is that there is an established 4-phase approach that any medical device company can quickly implement in order to accelerate growth:
- The Idea Phase
- The Research Phase
- The Development Phase
- The Commercial Phase
Following these steps will put you well ahead of the game when you receive FDA approval or FDA clearance.
What is a Medical Device?
Before we explore the four phases, we should define what a medical device is and isn't. As defined by the FDA, a product is a medical device if:
it is a device intended to diagnose, cure, mitigate, treat, or prevent disease OR it is a device intended to affect the structure or any function of the human body AND does not achieve any of its primary intended purposes through chemical action within the body AND is not dependent upon being metabolized for the achievement of any of its primary intended purposes
Medical Devices not only make up a sizable portion of the FDA regulated products, they also account for the most diversity among products.
As we explore the four phases of developing your quality management system, you'll understand how different medical devices have different requirements. In other words, there is no such thing as a generic QMS for Medical Devices.
Each unique device carries its own level of risk--from low-risk (e.g., tongue depressors and surgical gloves) to medium-risk (e.g., vitro diagnostic kits, CT Scanners, and surgical robots) to high-risk (e.g., wireless implantable devices and heart valves)-- and as a result, a corresponding set of QMS requirements.
The Idea Phase: How Quality Affects Medical Device Ideation
Once you've outlined a concept for a medical device, you'll kick off the Idea Phase.
The most important thing you can do, especially if this is your first device, is become knowledgeable about the industry and its regulatory requirements. Countless promising products never make it to market because their inventor/company didn't understand the criticality of taking some simple steps to ensure success.
Below are some actionable tasks to help you become knowledgeable and ideate:
Research the Medical Device industry
Understand FDA-defined product classifications and learn how risk categories are linked to the regulatory submission pathways of Exempt / 510k / PMA / De Novo. Specifically, find out the differences between clinical trials, non-clinical studies, and simulated use testing. Then, take that knowledge and apply it to your product.
Find Similar Products and Look for "Predicate Devices"
You may already know which of your competitors already have clearance or approval from the FDA. Start with them. Research their product claims and compare features, functions, and clinical value to what your product will offer. If they are similar, look up the competitor's product on the FDA's website and try to find their product code. This is a code the FDA assigns to a group of similar products.
In the event you are able to find "substantial equivalence" (SE) between your product and theirs, your regulatory submission process will be much easier. The FDA refers to a product already on the market that is substantially equivalent to yours as a predicate device. A competitor's predicate device is what you can leverage in your own submission to the FDA.
Read the Regulations
No one gets excited about reading regulations, however, skipping this step will cost you a great deal of time and money in the long run. Regulations lay out the minimum requirements for organizing your company, designing your products, and detail what you need to do to stay in a state of control. Even if you plan to get regulatory experts to help you, nothing beats a firsthand understanding of the requirements.
Read Warning Letters
This may seem like an odd step, but it's an opportunity to evaluate the areas of particular focus for the FDA. If you read enough Warning Letters, you will begin to see patterns, which will help you avoid the mistakes of others.
Set Realistic Expectations & Timelines
Estimating expectations and timelines is one of the most important steps of all. Far too often, small companies ask for help too late in the process to meet their original commercialization date, simply because they don't understand the lengthy timelines required to get through all phases. For example, a Traditional 510(k) submission (the most common) takes approximately 176 days to complete. A Premarket Approval Application (PMA) takes around 352 – 532 days to complete.
If you haven't started planning early enough and haven't built-in these timelines, your market launch will be delayed, costing you time to market and money. And then you'll have to explain this to your early investors.
The Research Phase: What Role Quality Plays in Research
In the Research Phase, you will refine the details for your step-by-step approach to a successful launch.
As you determine where you want to market your device with short-term and mid-term goals, you will refine your understanding of your product as it relates to safety and risk. This will then help you create a development model to use, and you will identify the components of your initial Quality system.
Let's dive into the actionable tasks critical to the Research Phase.
Develop a Marketing Plan and a Regulatory Plan
Having a marketing plan is an essential step towards understanding the regulatory requirements necessary to market your product in different countries. Without a well-thought-out market strategy, your regulatory plan will be woefully inadequate. It will likely cause delays and rework once you begin development.
Every country has its own regulatory pathways to get products to market. If you haven't accounted for these early in the process, you may not have what you need when entering that particular market. Much of the non-clinical & clinical data is the same; however, necessary documentation varies widely from country to country.
With a good plan, much of this can be set up for future implementation without undue work. Without it, you will find yourself constantly reproducing evidence instead of leveraging what you already have.
Define Submission Requirements
Just within the United States, there are a number of submission pathways. If you add in other countries, it can become quite complex. Safety and risk classifications may differ from country to country, along with their submission requirements. Selecting the correct submission pathway(s) depends on a number of factors. It is critical to engage experts here to ensure you have the correct submission pathway and any special requirements for the type of device you're making.
Did you know that there are three (3) types of 510(k) submissions? In addition to the 510(k)'s, there's the PMA process and the De Novo process. In Europe, there is a special process to obtain a "CE Mark." Without it, you cannot sell in Europe. These are just a few of the things you'll need to consider as you refine and develop the requirements to get your product on the market.
Determine your Development Model
There are lots of development models to choose from. The traditional Waterfall Model is the most common and easiest to understand for small companies; however, if your device is software, a Software Development Life Cycle (SDLC) model coupled with the V-Model for validation might be a better choice.
Another popular model is called AGILE Methodology. Although this is a popular method for fast prototyping, it is less effective in demonstrating compliance to all of the documentation requirements for design and development. Other models include SDM or Spiral Method, Evolutionary Prototyping Model, and Iterative and Incremental Methods similar to AGILE. For small companies not highly skilled in some of these more complex methodologies, we usually recommend sticking with the Waterfall Model or, for software, an SDLC/V-Model approach.
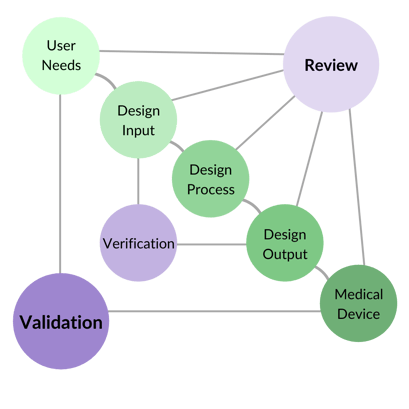
These models have a lower learning curve and are recognized by global regulatory authorities. Regardless of which one you choose, regulatory requirements need to be built into it. In the US, 21 CFR Part 820.30 defines those Design Control requirements. For the European Union, they are defined in ISO 13485:2016 Section 7.3.
In addition, Product Risk Management Standard requirements need to be implemented as part of the full life cycle for your device. These requirements can be found in ISO 14971:2019.
Define your Initial Quality Management System
At this stage, you certainly don't have to have a fully developed QMS in place, however, you should implement some foundational elements for your system to support the operations that you will have shortly.
A good way to identify which elements you need to implement is to review the 7 Subsystems of Quality and ask yourself: which of these apply to me now? Then, take those elements and implement processes and procedures around them.
The Development Phase: When to Develop Your Quality "Core"
In this phase, plans get put in motion and things get done. You'll implement the foundational processes for your quality management system ("The Core"). You'll fully develop your product, generate appropriate records for your market sector, and submit all necessary records in order to finally launch your product.
Let's dive into the actionable tasks critical to the Development Phase.
Implement "The Core" Processes for the QMS
At this stage, you should implement a number of foundational processes to support upcoming operations. The most critical element is that everything in a regulated environment must remain in a State of Control at all times.
For example, all documents, and the changes you make during design and development phases, must be controlled. For this, you will need to establish a Document Control System and a Change Control System.
Another component is people. In order to maintain a State of Control, people need to be trained and competent to perform work. For this, you'll need to establish an appropriate Training System.
Next, you'll need to establish a fully compliant Product Development Process that implements your chosen model in a way that incorporates all regulatory requirements necessary for Design Control.
Last but not least, you'll need to establish standard operating procedures and processes for the various suppliers and partners who will handle some of your outsourced processes (including contract manufacturing). Global regulatory requirements stipulate that you must have controls in place to ensure those suppliers are competent to provide the goods and services you require.
Let's take a deeper dive into each of these core processes.
Document, Record, and Change Controls for your Medical Device
All documents and records affecting the product or the quality system need to be approved and version/revision controlled. Each document has a life cycle that needs to be maintained. Each record provides evidence used to determine if the company followed its processes.
Records are required to be maintained for a period of time depending on the product's expected life. Changes to documents or product requirements also need to be controlled in a way so that any changes can be traced. All of these requirements start when a company begins developing a product. These processes are foundational or "core" because you really can't do much work in a regulated environment without them.
Ensure you have a Training Process
Training and competence in a regulated environment is not just something we say…we have to prove that the right people are performing the right jobs. Implementing an effective training program is a foundational or "core" process simply because no one can perform within a regulated environment without first proving that they have been trained and are competent to do the job.
Training needs to be conducted and documented before documents can become effective for the organization. Additionally, training needs to be conducted and documented for the design and development team before contributing to the design of a new product. Everything is affected by how effectively training remains in a State of Control.
Product Development Process / Design Controls
In the research phase, you decided on what general development model to use. That general model must be supplied with all required design control elements and then implemented. The development team needs to be trained on how to use it and what's required to be documented throughout the development process.
Supplier Controls
Most new medical device companies outsource some of their more critical operations, including sterilization, software development, critical custom parts, raw materials, and even the entire manufacturing process. But remember, outsourced or not, the company that owns the device is still 100% accountable and responsible for all processes and operations!
US and international regulations require a significant level of oversight of these suppliers, including a qualification process to approve them. The process of qualifying suppliers includes ongoing monitoring of their performance with consequences for non-fulfillment of their obligations and periodic re-qualification.
Throughout the development process, you will be making decisions on suppliers. You'll need to have this process in place early in order to have enough time to properly qualify suppliers.
Complete Submission Documentation
Whichever market(s) you enter, there will be regulatory requirements that must be met before you can offer your products for sale. If you thoroughly researched the required pathways, you already know what documentation you need and the general timelines to achieve clearance, approval, certification or licensing.
Now's the time to execute, gather, organize and submit that documentation. Once everything is submitted, there's a bit of a waiting game before there's more to do on that front. But that doesn't mean there's nothing to do! It's a great time to qualify any contract manufacturers, create detailed manufacturing instructions, perform process validation, and finish building out your Quality Management System.
The Commercialization Phase: Quality as a Continuous Process
In the Development Phase, we ended with the delivery of the submission documentation to the regulatory authorities. There may be some back and forth during the process as regulators review your submission, but once that's done to their satisfaction, you will finally get clearance or approval to put your device on the market. You will now enter the Commercial Phase.
In this phase, there are lots of things to do. First and foremost, you need to complete building out all of the required elements of your quality management system. This includes officially qualifying your suppliers, implementing all manufacturing specs, scaling-up from development to production, ensuring that you have quality oversight built into your processes, and maintaining your system.
Let's dive into the actionable tasks that must be accomplished before you can ship your first product.
Implement the rest of the QMS
In the Development stage, you implemented the foundational processes necessary to develop your product, but many more requirements need to be built into the QMS before it is considered compliant and in a State of Control. Remember, all of your processes need to be in place and validated where necessary, before you can ship product to end customers.
Here they are:
Management Responsibility
Executive Management is ultimately responsible for the safety of the product and for ensuring that the quality system is established, maintained, and compliant. Executive Management consists of the CEO or President and the senior executives who report to her/him.
Executive Management is required to review the effectiveness of the quality system and the product's status in the field at regular intervals. They are also responsible for ensuring that there are enough qualified people to manage the QMS and that the organization is structured to eliminate conflicts of interest and provide the appropriate authority to act. The executive team is also held accountable to ensure the training system is adequately maintained and that personnel are trained to perform their responsibilities.
Another important responsibility is to ensure that the organization executes internal quality audits according to a documented schedule. "Management Responsibility" is very vulnerable for small, emerging companies simply because they neglect to connect these senior executives to real responsibilities within the day-to-day quality management system. Remember, these audits are not optional. They are required by the regulations and standards governing this industry.
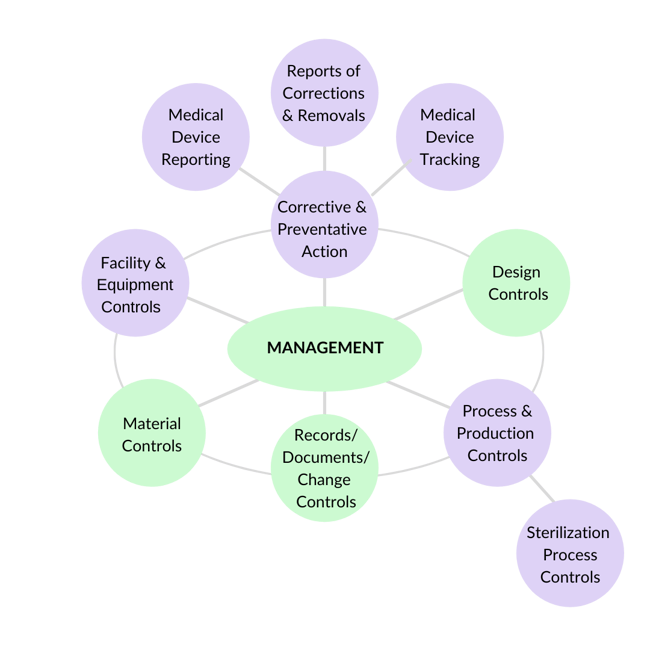
Corrective & Preventive Action (CAPA)
Many people think that CAPA is one singular process. The truth is that it is a group of processes that are all focused on one thing: monitoring the performance of products in the field and monitoring the health of the QMS. This area is also where the organization will take specific actions when things don't go the way they should. Some issues that fall under the "CAPA" area are things like analysis of data and metrics, feedback, complaint handling, reporting of adverse events to regulatory authorities, non-conforming product, rework, failure investigations, recalls & advisory notices, and, of course, corrective actions and preventive actions.
Each of these areas is different from a process point of view, so we have to create processes for each.Facility & Equipment Controls
If you are manufacturing your own product, you need to spend some time thinking about and implementing proper facility controls appropriate for your product.
Usually, renting office space in a strip mall doesn't work well for manufacturing medical devices. Is the building designed so that product flows through logically, so pieces and parts don't get mixed up? Have pest control measures been implemented? Is your product temperature or humidity sensitive? Will your product be sterilized? Are there contamination concerns? Is your product manufactured using equipment that needs to be qualified and maintained? Is your product sensitive to electrostatic discharge?
The answers to these questions will drive how many controls you need to put in place. If you are using a contract manufacturer, you are still 100% responsible for ensuring that your product is being manufactured in a facility that is compliant with these requirements.
Production & Process Controls
If you are manufacturing your own product, you need to translate the design and product specifications into manufacturing instructions. You'll also need to implement acceptance criteria for anything that could cause the product to fail in the field and implement inspection and testing points to ensure the product meets these criteria before shipping to customers.
Any automated processes also need to be validated if you cannot fully verify the output. Again, if you are using a contract manufacturer, you are 100% responsible for everything they do, so you need to qualify, monitor, and manage them.
Material Controls
This group of procedures and processes is all about how you handle, store, distribute, install (if required), and service (if required) not only your finished device, but also any raw materials, components, and sub-assemblies that you purchase from others. These processes ensure that materials and products are not damaged and are properly installed and serviced prior to use.
They also ensure that things are organized to prevent mixing-up materials, and that everything is identified and can be traced. Distribution records are extremely important if you ever have to recall the product from the field due to a problem.
Avoid the Quality Management System Kitchen Sink Scenario
With a well-thought-out foundational QMS built around simplicity and the proper requirements for your products and business model, you will avoid the painful "kitchen sink" scenario that so many small companies experience. This is when you throw everything imaginable into the QMS, whether you need it or not.
Small companies that don't have a plan for their QMS often end up buying a generic, cookie-cutter QMS off the internet at the last minute in a desperate attempt to have one in place. The trouble with these generic "quality-in-a-box" products is that they always take a "kitchen sink" approach to cover every possible situation. In the end, instead of making things simple and easy, these products complicate the process and end up doing the exact opposite due to the extreme diversity of medical device products on the market.
If you follow the advice in these four phases, you will find that getting your product on the market with a compliant and well-defined quality management system is less stressful than you might think.
Below is a quick summary of the four Phases to help you remember:
- The Idea Phase: Become knowledgeable and learn the price to pay
- Research Phase: Plan and define your scope
- Development Phase: Build "The Core"
- Commercial Phase: Execute!
As a final thought, it's important that a knowledgeable quality and regulatory compliance professional is involved in creating your QMS. Whether you hire them into the company or outsource, be sure they are qualified to do the work; otherwise, you may end up spending more money and time fixing their mistakes.
*An earlier version of some of this material was published on MBC & Affiliates, Inc. (MBCA)'s website.
About Melita Ball
Melita Ball is a Regulatory & Quality Systems professional with 28 years' experience in Life Sciences. She is the CEO and Principal Consultant at MBC & Affiliates, Inc. (MBCA). She has particular expertise in establishing compliant quality systems and leading large and small teams on remediation projects. Melita is considered an expert in global regulatory compliance requirements including: ISO 13485:2016, ISO 14971:2012, PMDA MO 169, BGMP 16/2013, TGA Therapeutic Goods (Medical Device) Regulations 2002, EU 2017/745 (New MDR), 21 CFR Parts 11, 801, 803, 806, 809, 810, 820, and MDSAP.
She is a recognized expert in providing consulting services for life science companies in the areas of Internal & Supplier Quality System Auditing, FDA 483 and Warning Letter mitigation, GxP Training, Medical Device Auditor Training, Purchasing & Supplier Controls, Software Validation (including electronic records & electronic signatures), Business Process Re-engineering, Design Controls, Management Controls, Complaint Handling systems, CAPA, Document Controls, Record Controls, and Change Controls. Melita is experienced in establishing and leading consulting teams through large projects to achieve successful outcomes for her clients.